在生物制品的开发过程和获得批准后,生产工艺的变更是不可避免的。变更的原因包括改进生产工艺、增加规模、提高产品稳定性,以及根据法规要求进行变更。
当生产工艺发生变更时,产品的质量、安全性和有效性都有可能受到影响,这就需要生产商总体评估产品的有关质量特性,以证明该变更没有对原液及制剂的安全性和有效性产生不利影响,从而成功得到监管机构的认同与批准。
可比性研究的法规要求
可比性研究是生物制品药学变更评价的基础和成功的关键。为了使企业在管理工艺变更时能够得到更好的指导,包括与监管机构更有效地沟通等,全世界的药品监管机构颁布了几个不同的法规指南,包括:
● ICH: Q5E: Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process
● FDA: Comparability Protocols for Post-approval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA
● EMA: Guideline on Comparability of Biotechnology-derived Medicinal Products after a Change in the Manufacturing Process
国家食品药品监管总局发布《生物类似药研发与评价技术指导原则》,对可比性研究的开展也具有十分重要的指导意义。
什么是可比性研究?
变更的目的是通过收集和评估相关数据,确定生产工艺的变更是否对产品产生任何不良的影响,确保生产工艺变更后产品的质量、安全和有效性。
而可比性研究则是通过评估可能影响产品安全性和有效性的质量之间的差异,来确定使用变更前产品进行的非临床研究和临床研究是否与变更后的产品依然相关的研究。理想情况下,可以确认变更前后产品质量具有可比性,但是如果无法证明,或者在可比性研究中发现影响产品安全性和有效性的重大差异,则需要额外的非临床或/和临床桥接实验以证明可比性。
具有可比性,并不意味着变更前和变更后的产品的质量特性一定要相同,但产品的安全性、鉴定、纯度和活性等应高度相似,并且能以现有知识充分预测,以确保质量特性上的任何差别对药物制剂的安全性或有效性不会产生不利影响。
风险评估
工艺变更的类型涉及从微小变更到重大变更。对于微小变更,不能获得可比性产品的风险很低;而重大变更也有不能获得具有可比性产品的风险。为了确定是否需要进行可比性研究,如果需要,在确定可比性研究的深入程度上,一般会采用ICH Q9中所概述的风险评估程序进行评估。
风险评估有助于确定可比性的研究范围、协助批次选择、分析方法以及需要进行哪些研究(如扩展表征、强制降解等)。风险评估应该侧重于产品及其特性,Table 1中列出了一些可比性风险及其研究内容的要求。
Table 1.可比性风险及研究内容

可比性的研究内容
批次的选择
在决定可比性研究包含的批次数量时,需要考虑产品开发阶段、变更类型以及对工艺和产品的理解。尽管使用多个批次可以证明过程的稳健,但它可能不可行或没有必要,尤其是对于处于开发阶段的项目。
可比性研究的样品在指导文件中对研究批次的描述并不清晰。FDA建议比较平行检测多个单独的产品批次;而ICH Q5E则规定对于已上市的产品,应对恰当批次进行变更后产品的分析,以证明工艺的一致性。
对于重大变更,一般选择变更后≥3批连续的商业化规模的样品;而中等变更则选择变更后3批连续的商业化规模的样品。微小变更可以用较少的变更后批次进行研究,一般选择≥1批连续的商业化规模样品。
如果要减少可比性研究的批次(采用括号法、矩阵法)等或者缩小研究规模(扩大规模的变更除外),应在科学和风险评估的基础上,提供充分的依据。
可接受标准
应制定前瞻性的可接受标准。可比性研究的可接受标准不等于质量标准。可接受标准要根据工艺和产品质量的历史数据设定,排除任何数据都应有充分的理由。设定的可接受标准不能低于质量标准,除非证明是合理的。根据研究方法的性质,可比性研究验收标准可分为定量标准和定性标准,定量标准要满足范围要求,定性标准则看图谱比较。
评估变更后产品的可接受标准时,考虑ICH Q6B中关于质量标准设定的基本原则,如变更对已验证的生产工艺、特性鉴定研究结果、批分析数据、稳定性数据、非临床和临床经验的影响。
可比性研究可接受标准的制定可以参考Table 2。
Table 2.可比性研究可接受标准制定参考表
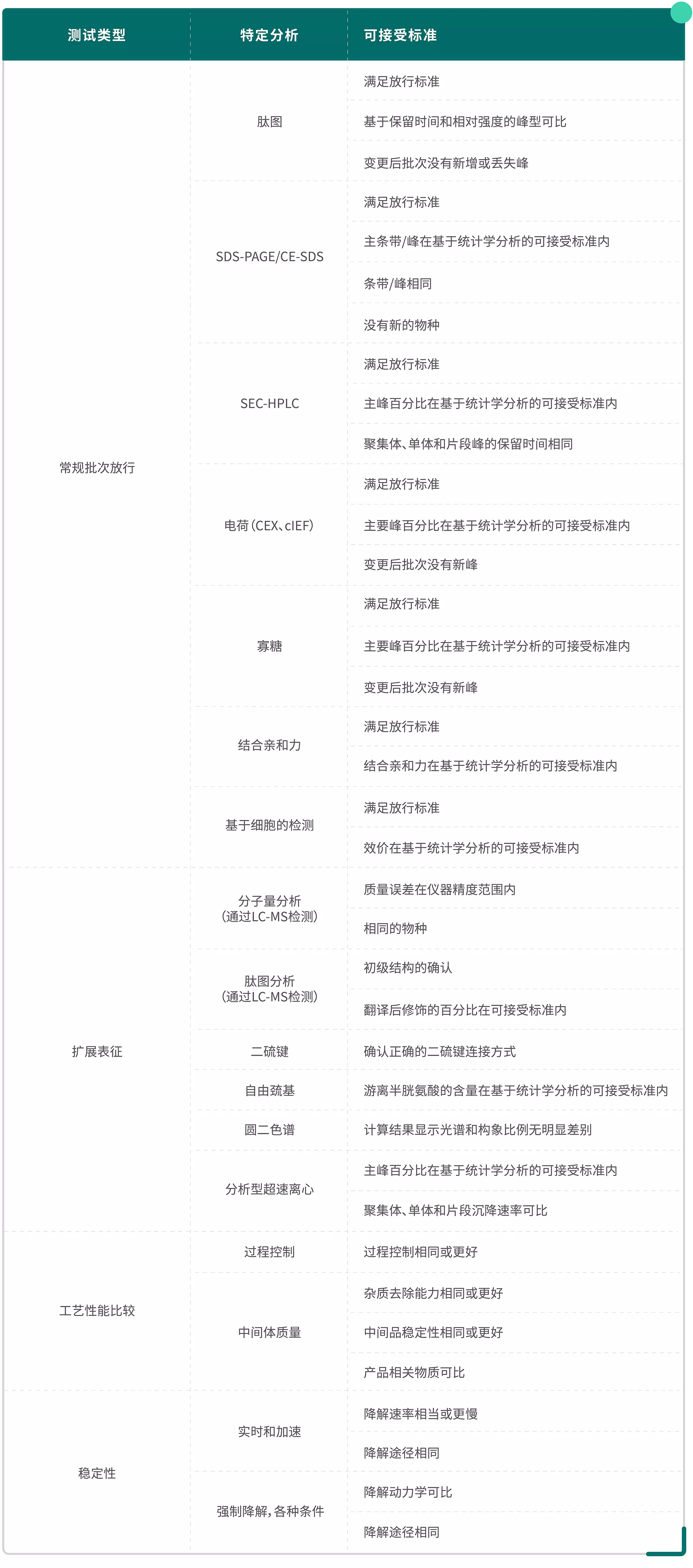
工艺性能的比较
工艺可比性研究主要是证明工艺的稳健性和批间的一致性,使变更前后工艺和中间产物具有可比性,且变更后的工艺控制能力不低于变更前。
进行工艺评估应考虑诸多因素,如工艺步骤、工艺参数、过程控制结果与历史数据的比较,以及额外工艺过程控制参数的比较(必要时)等。如果可证明单一变更对后续工艺及产物无影响,验证可以限制在被影响的工艺步骤内进行;如果对后续产生影响,应同时对后续步骤进行验证。
应关注变更前后生产工艺对有关物质、杂质和外源因子的去除能力的可比性,必要时,应开展病毒去除/灭活效果验证,开展中间品保存研究,过滤膜和层析柱使用寿命研究,以及开展一次性使用产品的可提取物及可浸出物对产品质量的影响验证等。
质量的比较
用于可比性研究的质量对比数据通常来自常规批次放行检测和扩展表征,将变更后的产品质量与历史数据进行比较。当原液的变更影响到制剂时,应同时展开原液和制剂质量的可比性研究。
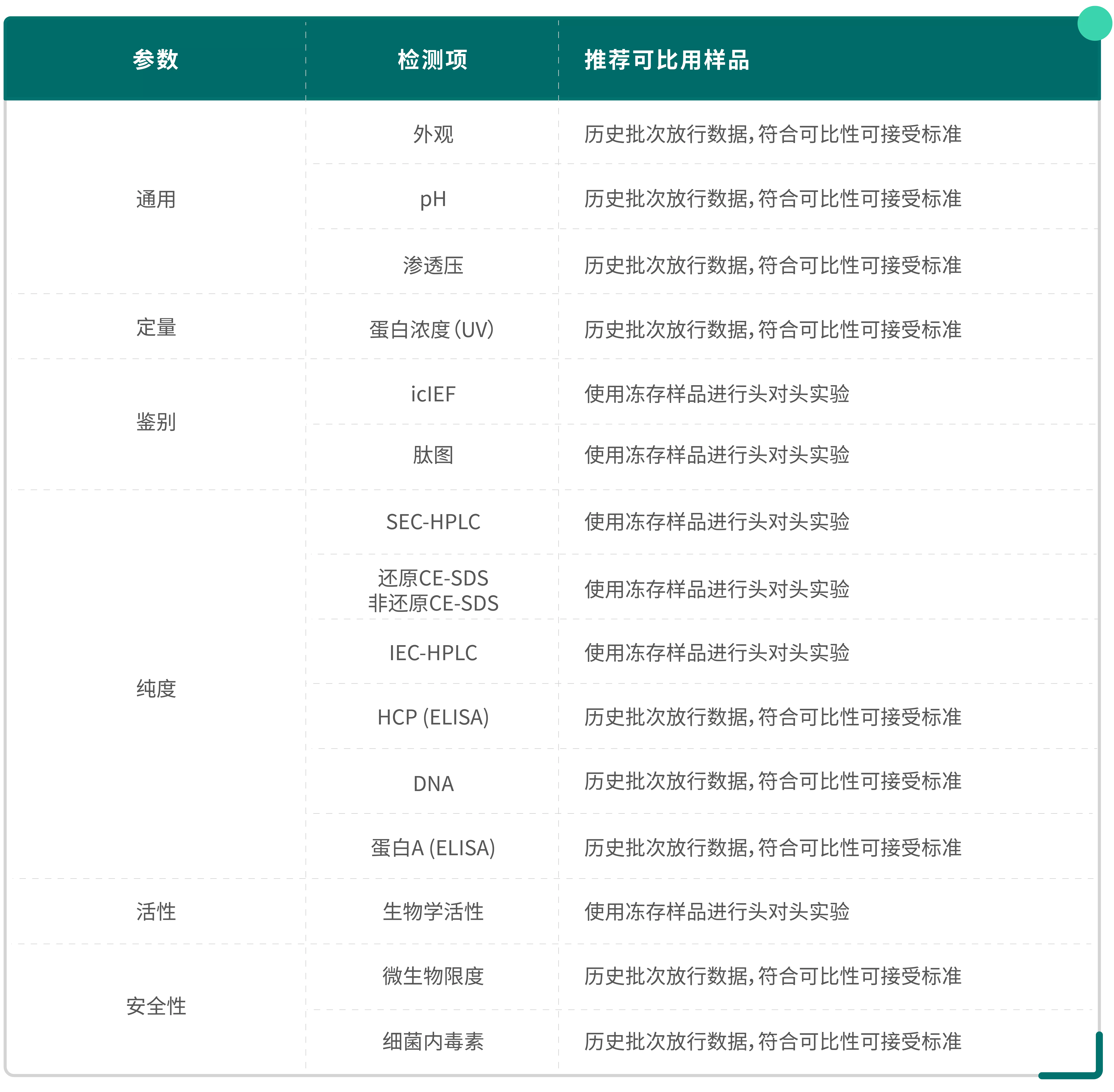
Table 3中列出了常用的单抗原液放行检测项目。
Table 3.常用单抗原液放行检测项目
除了常规的放行检测之外,扩展表征分析测试也会进行,以便能够更全面地检测产品的变化对安全性和有效性的影响,或者获得对变更后产品发生的分子结构变化的更细节的评估。
常用的扩展表征分析方法主要用于分析产品的初级结构、二级结构和更高级的结构,电荷异构体、糖基化、氧化和纯度。由于这些分析方法要比常规用于产品检测放行的方法更复杂,且没有足够的历史数据用于变更前后产品的对比分析,因此通常采用头对头的对比分析。
稳定性的比较
稳定性研究能够检测出那些通过结构确证研究不能检测到的细微差异。同样地,当原液的变更影响到制剂时,应同时展开原液和制剂的稳定性研究。稳定性方案应当包括加速条件和实时条件下的稳定性研究,如果可以,还需要包括强制降解研究。
ICH Q5E强调“必要的稳定性数据,包括加速或强制降解条件下的稳定性数据,以更深刻理解产品降解途径上的潜在差异,以及由此导致的产品相关物质和产品相关杂质的潜在差异”。
加速,尤其是强制降解稳定性研究,通过产生相对较高的降解水平,可以在短时间内对变更前后的批次进行头对头的比较,能够更好地建立产品降解趋势,比较降解产物及降解动力学。
早期进行的可比性研究可能不需要强制降解研究。
可比性桥接研究
产品可比性的确定以质量特性研究为基础,当质量数据对确定可比性不充分时,可以适当地增加非临床和临床研究以获得补充证据。
ICH Q5E中涉及的非临床和临床研究包含药代动力学(PK)研究、药效动力学(PD)研究、临床有效性研究、特异的安全性研究、免疫原性研究和药物警戒研究等。
非临床和临床研究的范围和类型要基于不同因素进行具体问题具体分析。主要考虑的因素有以下三点:一是质量研究结果,如新杂质可能需要进一步的毒理学研究。二是对产品的认知水平,包括产品质量属性与安全性及有效性的关系,产品作用方式和作用位点等。三是产品现有的非临床和临床数据,如长期给药比短期给药对某种差异可能带来的风险会更高;皮下注射比静脉注射通常更易引起免疫原性等。
可比性的研究结果
(1)基于产品质量特性研究的数据满足预定义的可接受标准,这种情况不需要再进行额外的研究。
(2)变更前后的产品质量特性研究数据有差异,如果生产商能够基于对分子的理解和科学知识,可以证明该差异对安全性和有效性没有不利影响,则即使变更前后的质量属性超出预定义的可接受标准,也可能不需要再进行额外的研究。
(3)仅基于质量数据的评估无法建立可比性,则需要进一步进行非临床或/和临床研究证明变更前后的可比性。
(4)如果所有的研究数据都无法证明变更前后产品的可比性,则要么将变更后的产品作为一个新的产品进行开发,要么不再选择执行变更后的工艺。
参考文献
[1] ICH Q5E Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process
[2] FDA Comparability Protocols for Post-approval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA
[3] EMA Guideline on Comparability of Biotechnology-derived Medicinal Products after a Change in the Manufacturing Process
[4] The Development of Therapeutic Monoclonal Antibody Products: A Comprehensive Guide to CMC Activities from Clone to Clinic
[5] NMPA《已上市生物制品药学变更研究技术指导原则(试行)》
[6] NMPA《临床试验期间生物制品药学研究和变更技术指导原则(试行)》
[7] NMPA《预防用疫苗临床可比性研究技术指导原则》